発作性夜間ヘモグロビン尿症
1 琉球大学医学部附属病院骨髄移植センター
2 琉球大学医学部第2内科
友寄 毅昭1、2
仲地 佐和子2、西田 紀子1、2、奥平 多恵子1、2、百名 伸之1、益崎 裕章2
【要旨】
発作性夜間ヘモグロビン尿症(paroxysmal nocturnal hemoglobinuria: PNH)は 後天性溶血性貧血で、早朝のヘモグロビン尿が特徴である。病因はX 染色体に存在 するglycosyl-phosphatidylinositol(GPI)アンカーの生合成を支配するPIG-A 遺伝子の変異である。GPI アンカー型蛋白は赤血球のみでなく全血球に及んで欠損 しているので症状は多彩で、溶血発作にとどまらず、汎血球減少や血栓症も合併す る。診断には従来から行われている砂糖水試験、Ham 試験があるが、CD55、 CD59 欠損PNH 血球をフローサイトメトリー法で検出する方法が感度、特異度と も高い。治療は鉄欠乏に対して少量の鉄剤、重度の貧血では濃厚赤血球の輸血を行 う対症療法が主体である。溶血抑制目的にプレドニゾロンを用いることもあるが、 溶血や血栓が強い場合には補体阻害作用のあるエクリズマブがより有効である。
はじめに
発作性夜間ヘモグロビン尿症(paroxysmal nocturnal hemoglobinuria: PNH)は後天性 溶血性貧血で、夜間に血管内溶血をおこし、早 朝の褐色尿(ヘモグロビン尿)を認めることが 特徴でこの病名がある。補体に対して異常な感 受性を獲得した赤血球(PNH 赤血球)が補体 の攻撃により血管内溶血を起こす。しかし、 PNH の本態である補体に弱い特徴は赤血球に とどまらず、白血球や血小板の全血球に及ぶ。 そのため症状が多彩で、溶血発作だけでなく、 汎血球減少や血栓症、慢性腎臓病を合併し死に 至る。また、軽症例では積極的に疑わないと診 断がつかないことがあり注意を要する。
A. 疫学と病態
PNH の推定有病率は100万人に1 〜 5 人と され、平成10 年の厚労省の疫学調査(大野班) で本邦の推定有病者数は430 人であった。男女 比はほぼ1:1 で、診断時年齢分布は20 〜 60 歳 代に多く、まんべんなく発症する。病因はX 染 色体に存在するglycosyl-phosphatidylinositol (GPI)アンカーの生合成を支配するPIG-A 遺 伝子の変異である1、2)。GPI アンカー型膜蛋白 とはGPI と呼ばれる糖脂質をアンカー(錨:い かり)として、血球細胞膜につなぎ止められて いる蛋白のことである。ヒトにはおよそ150 種 類のGPI アンカー型蛋白が存在している(表 1)が、合成障害による不完全なGPI アンカー ではGPI アンカー型膜蛋白は細胞表面にとどま ることができない(図1)。GPI アンカー型膜蛋 白の代表的な蛋白がCD55、CD59 であり、こ れらは補体制御因子といわれている。変異血球 膜表面におけるCD55、CD59 の補体制御因子の欠損により、自己の補体の攻撃を受けやすく なるため赤血球では血管内溶血を来す。病名の 由来となった早朝の肉眼的ヘモグロビン尿が起 こる機序としては夜間睡眠中にCO2や乳酸など が蓄積し、局所的に血液pH が酸性に傾くこと によって補体が活性化され、溶血が生じると考 えられている。補体が活性化する誘因としては 夜間以外に、感染症、妊娠、手術、ビタミンC 過剰摂取などがある。PNH ではこれら誘因に より一時的に補体の活性があると大量の溶血に より肉眼的なヘモグロビン尿を認める。
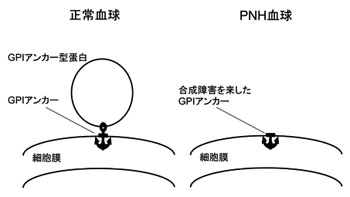
(脚注)PNH 血球ではPIG-A 変異によりGPI アンカーが合成 されないため、GPI アンカー型蛋白が細胞膜表面に結合でき ず欠損している。GPI: glycosylphosphatidylinositol
正常な細胞は、補体障害を回避できるよう に、可溶性補体制御因子であるC 1 I N H 、 C4BP、Factor H, Factor I などや補体制御膜 蛋白であるCD35、CD46、decay-accelerating facto(DAF; CD55)、protectin(CD59) により補体の攻撃から保護されている。しか し、G P I アンカー型膜蛋白であるC D 5 5 、 CD59 の補体制御膜蛋白を欠損するPNH 赤血 球は他の補体制御蛋白だけでは補体からの攻撃 を完全には防御できず、補体活性化を機に補体 の攻撃をうけ溶血する。実は、PNH の後天性 PIG-A 遺伝子変異は多能性幹細胞レベルでの 異常であり、顆粒球やリンパ球、血小板など他 の血球系統にも異常が及ぶ。また、GPI アンカ ーの合成障害により血球に欠失しているGPI ア ンカー型蛋白はCD55、CD59 の膜蛋白だけで なく、接着因子や酵素、血液型抗原などに関与 する蛋白にも及ぶ。遺伝子変異もフレイムシフ ト変異が半数以上を示すが、ミスセンス変異、 スプライス変異、ナンセンス変異などもあり、 GPI アンカー型蛋白は完全欠損から部分欠損ま である。PNH 患者では、GPI アンカー型蛋白 欠損の異常血球と正常血球が混在しており、異 常赤血球の割合が多いほど、特にGPI 完全欠 損型血球が多いほど症状も重篤化する。
B. 症状
PNH の症状には、疲労感、呼吸困難、腹痛、 嚥下障害、睡眠障害、QOL の低下、ヘモグロ ビン尿、勃起不全などがある。合併症として血 栓症、汎血球減少、肺高血圧症、慢性腎臓病 (CKD)などがある。GPI アンカー型蛋白の欠 損が多岐にわたれば、症状も多彩になると思わ れる。PNH 特有の溶血・血栓症状と汎血球減 少の二面性がある。病名の由来ともなっている 肉眼的ヘモグロビン尿は必ずしも全症例に認め られるわけではなく、診断時にヘモグロビン尿 を呈する症例は全体の1/3 といわれ、肉眼的血 尿がなくとも溶血性貧血があればPNH も念頭 において鑑別する必要がある。溶血以外に血小 板の機能異常も関与して腸間膜静脈や肝静脈、 門脈の血栓症を来たし、急な腹痛を訴えること もある。動脈系よりも静脈系の血栓症が多い。 本邦では血栓症の頻度は4 〜 10 %といわれ、 欧米の30 〜 40 %台と比べて少ないが、人種間 で頻度が違う理由はよくわかっていない。しか し、前述した原因不明な血栓症をみた場合に は、PNH も念頭に鑑別を進める必要がある。 好中球アルカリフォスファターゼ(NAP)、 CD14、CD16 などの欠損では好中球減少に加 えて好中球機能低下ももたらし、易感染性とな る。また、血小板減少による出血傾向も認め る。汎血球減少は診断時に約40 %認める。
C. 検査所見
通常、正球性正色素性貧血となる。しかし、 血管内溶血が長期にわたると、鉄欠乏を合併 し、小球性を呈する。網赤血球は一般に増加し ている。白血球、血小板数も減少し、汎血球減少を呈することが多い。NAP score は低値と なる。骨髄は正形成、過形成、低形成それぞれ が認められる。生化学検査では間接ビリルビン の上昇、LDH の上昇、ハプトグロビンの低下 を認める。中でもLDH の高値は著しく、2,000 IU/L を越える例もまれではない。尿ではヘモ ジデリン尿が特徴的である。
表1. PNH 血球で欠失しているGPI アンカー型蛋白

特殊検査としては、砂糖水試験、Ham 試験 (酸性化血清試験acidified serum test)、など がある。Ham 試験では溶血の程度がわかるが、 検査手技は面倒である。しかし、最近ではフロ ーサイトメトリー検査が標準的な検査になりつ つある。フローサイトメトリー法で血球膜上に おけるCD55、CD59 の発現により、GPI アン カー型蛋白の欠損血球を感度・特異度も高く検 査することができる。さらに、フローサイトメ トリー法では1 %未満のCD55、CD59 陰性血 球も検出することが可能である。PNH 血球を評 価すべき病態、疾患として1)ヘモグロビン尿、 2)クームス陰性血管内溶血性貧血(特にLDH 高値、鉄欠乏を伴う場合)、3)非典型的な静脈 血栓(Budd-Chiari 症候群、腸間膜や門脈の血 栓、脳血管)、4)再生不良性貧血、5)骨髄異 形成症候群(不応性貧血)、6)原因不明の嚥下 障害、血管内溶血を伴う腹痛などである3)。
D. 診断
典型的な症例であれば、病歴と症状で容易に PNH を推測できる。しかし、非典型例や病初 期では他疾患と類似しているため、診断までに 数年を要することがある。貧血、網赤血球の増 加、ハプトグロビン低値で溶血性貧血を疑う が、クームス試験陰性であればAIHA の可能性 は低くなり、家族歴や赤血球形態などで先天性 溶血性貧血を否定できれば診断確定のため砂糖 水試験やHam 試験の特殊検査を行う。また、 最近ではより感度、特異度も高いフローサイト メトリー法で解析することが多く、GPI アンカ ー型蛋白(CD55、CD59)欠損血球を1 %以 上認めれば、溶血所見が顕性化するといわれる のでこのようなPNH は古典的PNH(classical PNH)と診断してよい。また、再生不良性貧 血(aplastic anemia: AA)や骨髄異形成症候 群(myelodysplastic syndrome: MDS)と鑑別 を要する、またはAA とPNH を相互移行する 症例もあり、血球減少を伴うPNH を、骨髄不 全を伴うPNH(PNH in the setting of another specified bone marrow disorder)としてい る。この中にはとくに小児や若年PNH で認め るMDS が先行する例もある4)。さらに、一見 臨床的には問題にならないと思われるような 0.003 %以上というごくわずかなPNH 血球を AA では10 〜 50 %、MDS では10 〜 20 %に検 出される。微小PNH 血球が検出されるAA、 MDS では免疫抑制療法の反応性が良好という 特徴がある5)。溶血が明らかではないPNH を PNH-subclinical in the setting of another specified bone marrow disorder としてPNH を3 つの病型に分類している3)。
E. 治療
根本的治療としては造血幹細胞移植が唯一の 治療であるが、イタリアでは19 年間に26 例の PNH に対して同種移植を行い、10 年無病生存 率57 %、移植関連死は42 %の成績であった6)。 移植合併症のリスクがあり、血栓症や反復する 溶血発作、汎血球減少の程度によるが、同種移植は重症例に限定されることが多いだろう。貧 血に対しては造血能を促進する蛋白同化ステロ イドである酢酸メテノロン(プリモボラン(R)) が通常用いられる。溶血発作に対しては副腎皮 質ステロイドを用いる。ヘモグロビン尿による 鉄欠乏に対しては少量の鉄剤を投与する。ただ し、鉄剤は異常赤血球の産生が亢進し、溶血発 作の誘因にもなるので過量投与にならないよう 注意が必要である。貧血が重度であれば輸血を 行うが、補体を含む血漿成分は少ないので、濃 厚赤血球で十分なことが多く、従来推奨されて きた洗浄赤血球輸血は必ずしも必要ではない。 溶血発作が急激におこり、大量のヘモグロビン 尿が排出され、腎不全が危惧される場合には、 ハプトグロビン製剤の点滴を行う。血栓症対策 としてはワーファリンやヘパリンなどによる抗 凝固療法を行う。溶血に対して補体第5 成分 (C5)に対するヒト化モノクローナル抗体であ るエクリズマブ(ソリリス(R))は劇的な効果が あり、本邦では2010 年に承認された。補体活 性化経路には近位補体経路と終末補体経路があ るが、エクリズマブは近位補体活性に影響を与 えずに、補体C5 に高い親和性で結合すること により終末補体経路のみを阻害する。作用機序 から当然、エクリズマブは根本治療ではなく、 投与により溶血が抑えられる分、エクリズマブ 投与後のPNH 血球はむしろ増加する。終末補 体複合体は髄膜炎菌の除去にも働いているの で、エクリズマブは、髄膜炎感染症合併のリス クが生じる。投与前に髄膜炎菌ワクチン接種が 望ましい。ワクチン接種後も髄膜炎菌感染症の 合併に注意する。日本人を対象にした第II 相試 験であるAEGIS 臨床試験では溶血が87 %抑 制された7)。エクリズマブは新しい薬剤である が、終末補体経路を抑制することによる髄膜炎 菌を始めとする感染症の問題、高額医療となる 医療経済的な問題などがあり、PNH 患者全例 を対象とした薬剤ではない。溶血発作を繰り返 し輸血依存になる症例や、生命予後に関わる血 栓症を有する症例が適応になる。骨髄不全症 (汎血球減少)には効果がないので、その際は リスクを考慮して造血幹細胞移植を行う。
F. 予後
5 %は自然寛解するといわれている。古典的 PNH は慢性に経過し、比較的予後は良好で発 症/診断からの長期予後は本邦では平均生存期 間が32.1 年、50 %生存が25 年である8)。本邦 では重症感染症、出血、腎不全、MDS ・白血 病、血栓症が主な死因であるが、米国では血栓 症、重症感染症、出血、MDS ・白血病と血栓 症が最も多かった。血栓症を伴うと予後不良と なるが、エクリズマブの登場でさらに予後は改 善することが期待される。
終わりに
PNH は後天性PIG-A 遺伝子変異によるGPI アンカー欠損症であり、GPI アンカー型蛋白を 欠く疾患である。病因遺伝子も判明し、エクリズ マブという新規薬剤は出たが、未だ病態が不明な 点や安全に治癒をもたらす治療法の開発にはたど り着いておらず、今後の発展に期待したい。
文献
1)Hidaka M, et al. Impaired glycosylation of
glycoshlphosphatidylinositol-anchor synthesis in
paroxysmal nocturnal hemoglobinuria leucoytes.
Biochem Biophys Res Commun 191: 571-579, 1993.
2)Takeda J, et al. Deficiency of the GPI anchor caused
by a somatic mutation of the PIG-A gone in
paroxysmal nocturnal hemoglobinuria. Cell 73:703-
711, 1993.
3)Parker C, et al. Diagnosis and management of
paroxysmal nocturnal hemoglobinuria. Blood
106:3699-3709, 2005.
4)Ware RE, et al. Paroxysmal nocturnal hemoglobinuria
with onset in childhood and adolescence. New Engl J
Med 325:991-996, 1991.
5)Horikawa K, et al. Cyclosporine-responsive
pancytopenia and HLA class II alleles of a patient
with paroxysmal nocturnal hemoglobinuria. Int J
Hematol 63:165-166, 1996.
6)Santarone S, et al. Hamatopoietic stem cell
transplantation for paroxysmal nocturnal
hemoglobinuria: long-term results of a retrospective
study on behalf of the Gruppo Italiano Trapianto
Midollo Osseo (GITMO). Haematologica 95:983-
988, 2010.
7)Kanakura Y, et al. Safety and efficacy of the terminal
complement inhibitor eculizumab in Japanese patients
with paroxysmal nocturnal hemoglobinuria: the
AEGIS clinical trial. Int J Hematol 93:36-46, 2011.
8)Nishimura J, et al. Clinical course and flow
cytometric analysis of paroxysmal nocturnal
hemoglobinuria in the United States and Japan.
Medicina (Baltimore) 83:193-207, 2004.
Q U E S T I O N !
次の問題に対し、ハガキ(本巻末綴じ)でご回答いただいた方で6割(5問中3問)以上正解した方に、 日医生涯教育講座0.5単位、1カリキュラムコード(64.肉眼的血尿)を付与いたします。
問題
次の設問1)〜 5)に対して、○か×でお答え下さい。
- 1)発作性夜間ヘモグロビン尿症は血管外溶血である。
- 2)発作性夜間ヘモグロビン尿症は汎血球減少を来す。
- 3)発作性夜間ヘモグロビン尿症による血栓症は静脈系より動脈系血栓症が多い。
- 4)発作性夜間ヘモグロビン尿症では直接クームス試験が陽性となる。
- 5)発作性夜間ヘモグロビン尿症に対してエクリズマブを投与する際は髄膜炎菌感染症に注意する。
CORRECT ANSWER! 7月号(Vol.47)の正解
日常臨床における脂質異常症の取り組み:最近の考え方
問題
次の設問(1)〜(5)に対して、○か×でお答えください。
- (1)家族集積のある高HDL コレステロール血 症はすべて長寿家系とみなすことが出来る。
- (2)糖尿病患者では動脈硬化惹起性のリポ蛋白 が増加していることが少なくない。
- (3)人類の遺伝性脂質異常症の中で最も高頻度 な疾患は家族性複合型高脂血症(FCHL)で ある。
- (4)低HDLコレステロール血症の対処法とし て減量は有効な手段とは言えない。
- (5)甲状腺機能低下症ではしばしば高LDL コ レステロール血症を伴う。
正解 1.× 2.○ 3.○ 4.× 5.○